本教程参考。
引用文献:Chen, M., Bell, J.M., Shi, X. et al. A complete data processing workflow for cryo-ET and subtomogram averaging. Nat Methods 16, 1161–1168 (2019)
下载测试数据
教程使用的数据可以在EMPIAR数据库上下载:(4套Tilt serials)。
文件中header的pixel size存在错误。导入图像时应指定正确的值(2.62)。
导入文件
为项目创建一个新的空文件夹,并进入该文件夹,运行:
mkdir /path/to/testdir
cd /path/to/testdir
e2projectmanager.py
设置Workflow Mode为Tomo。
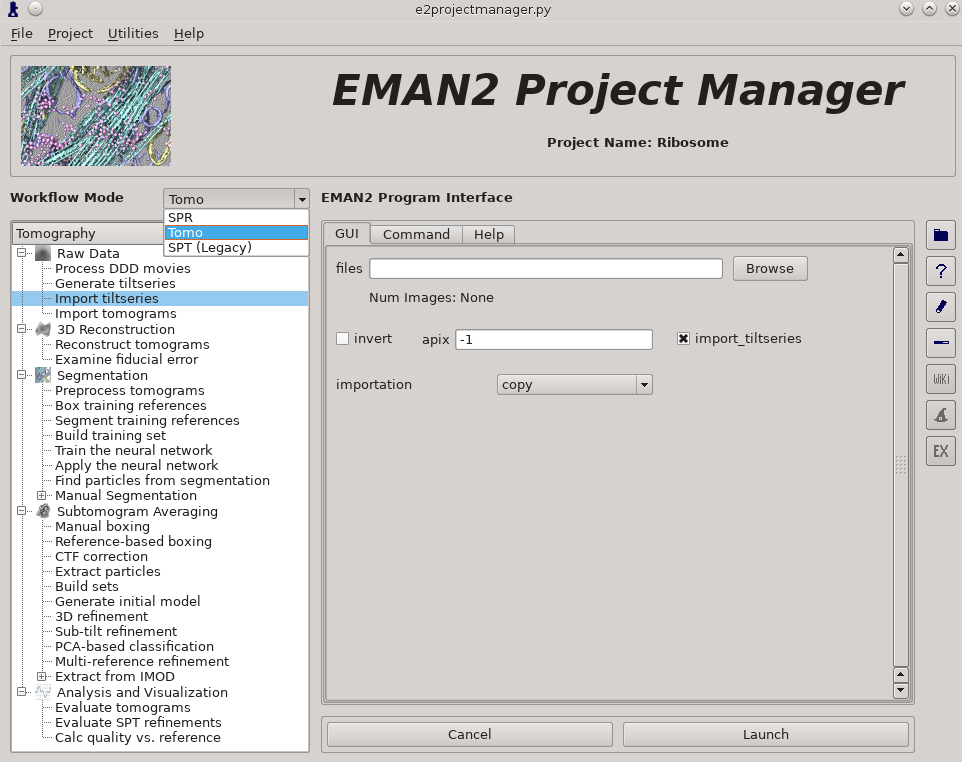
选择Raw Data > Import tiltseries。
-
在
apix框中输入正确的pixel size值,EMPIAR-10064这套数据为2.62。 -
点击
Browse并选择原始数据文件。文件名中不能包含任何空格或点(除了文件扩展名的最后一个点)。 -
设置好选项后,点击
Launch。-
如果从Motion corrected的Micrographs开始,选择
Raw Data > Generate tiltseries。可以选择属于一个tilt series的所有micrographs,逐个构建tilt series。也可以选择一个文件夹,将所有显微照片放进去,勾选guess并点击Launch。程序会根据文件的命名猜测哪些文件对应一个tilt series及其倾斜角度。对于使用较多的数据收集软件如SerialEM、EPU等生成的照片,大多数情况下都能正常识别。每个tilt series会创建一个合集(.lst文件)。 -
如果是已经做完Motion correction的Tilt series,也可以直接选择
Raw Data > Import tiltseries。
Tilt series对齐和tomogram重建
-
在EMAN2中,tilt series的对齐与tomogram重建是迭代进行的。tomogram通常不以原始大小重建,通常为1k x 1k或2k x 2k,通过命令行也能在原始4k重建,但是并不建议也不太需要,对于的sub-tomogram averaging,使用原始tilt series数据,在下采样的tomogram中挑选的颗粒坐标。tilt series是以原始大小对齐的。
对于教程中的tilt series:
选择3D Reconstruct > reconstruct tomogram:
-
勾选
alltiltseries,或者从tiltseries文件夹中选择tilt series -
这套数据是蛋白样品而非细胞,冰比较薄,可以勾选
correctrot -
tltstep= 2 -
clipz= 96(单位是1k下的pixel) -
点击
Launch -
当样品较厚时,重建中可能会看到一些网格状的图案。勾选
extrapad可以大大减少这些边界伪影。还有一个moretile选项可以进一步消除这些伪影。勾选这些选项可能会使重建过程更加消耗内存,并且速度可能减慢多达5倍。这些伪影不会影响subtomogram averaging结果。 -
对于冰层较薄的样品,比如这套教程数据的单颗粒样品,手动指定
clipz值以生成更薄的tomogram可能有所帮助。
CTF估计
选择3D Reconstruct > CTF estimation:
-
勾选
alltiltseries -
检查
voltage和cs是否和电镜收集参数一致,教程数据是300和2.7 -
前两个选项
dfrange和psrange表示要搜索的defocus范围和phase shift范围,格式为“开始,结束,步长”,比如“2,5,0.1”将从2到5 μm搜索defocus,步长为0.1 μm。相移同上,单位为度。 -
点击
Launch
此程序仅估计CTF参数,不会对整个tomogram进行校正。CTF校正在后面对每个颗粒的执行。此过程需要tilt series对齐期间确定的元数据,因此不能与其他软件包重建的tomogram一起使用。
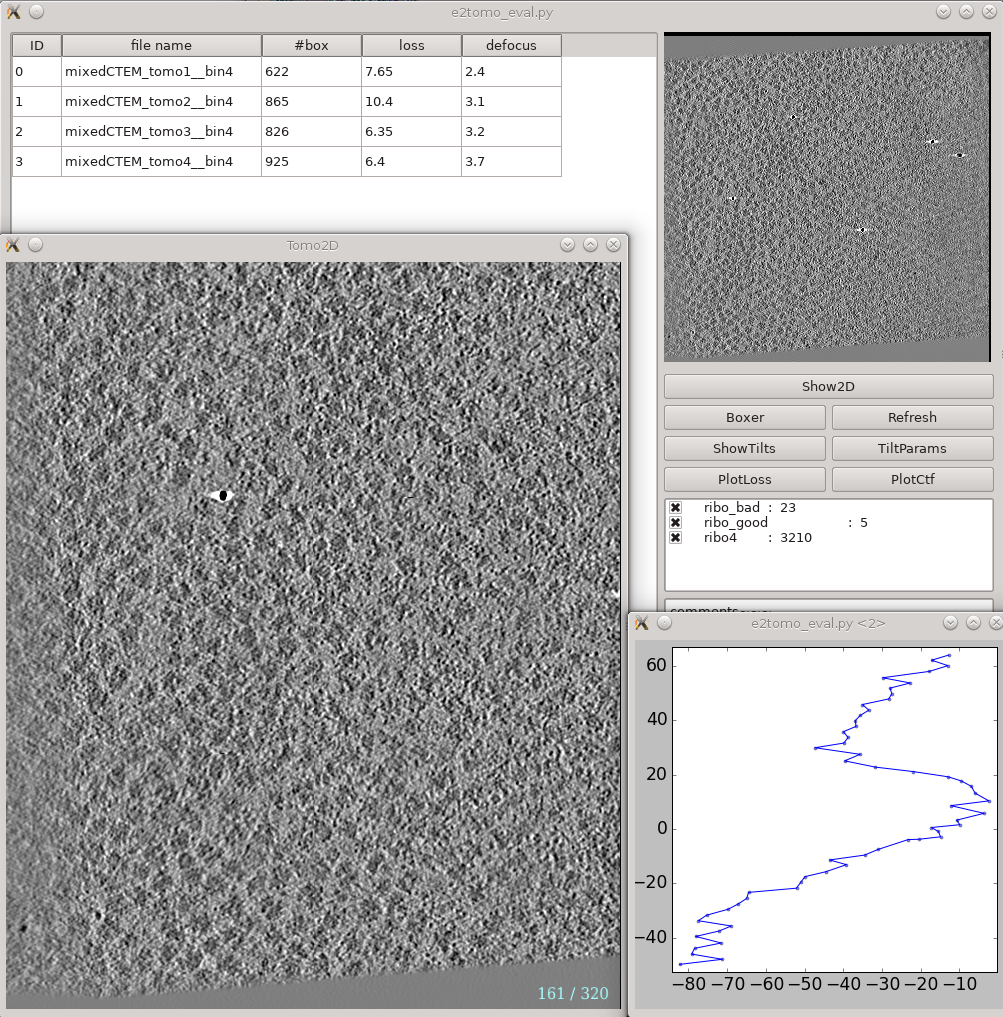
Tomogram评估
Analysis and Visualization > Evaluate tomograms可用于评估tilt series对齐和tomogram重建的质量。
颗粒挑选
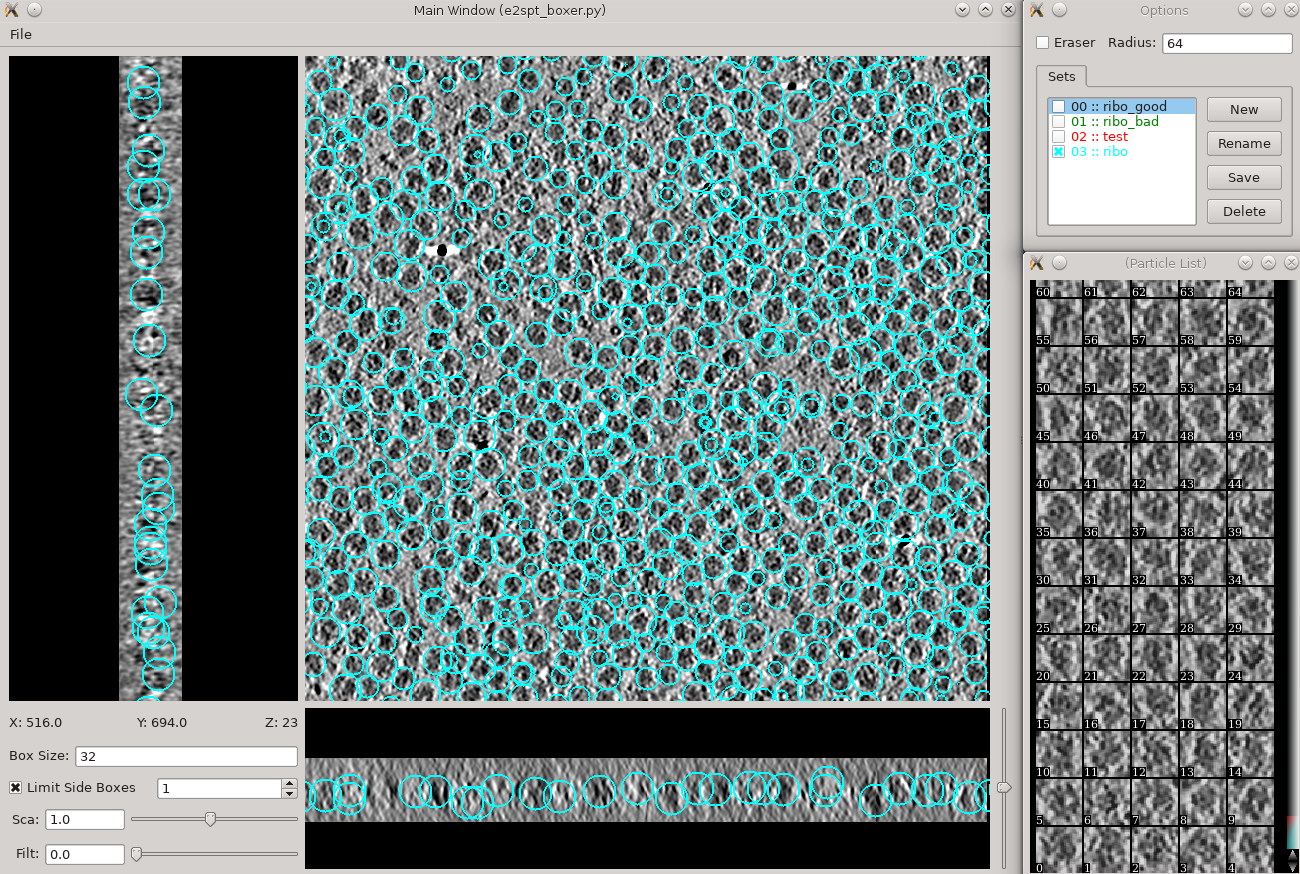
选择Subtomogram Averaging > Manual boxing :
-
rename填写一个标签,比如“initribo”,后续阶段用标签名字来指定颗粒集合。单个tomogram中可以有不同box类型,每种类型都有一个标签。如果在不同tomogram中选择相同的特征,请确保标签是一致的。 -
使用键盘上的~键和1键沿z轴滑动切片
-
左键点击添加一个box
-
按住Shift键点击左键删除box
-
Box大小可以在主窗口的左下角设置,对于核糖体使用48(unbin的大小为192)。
从一个tomogram中选择30-50个颗粒,然后关闭窗口,会自动保存的。那个Save键不需要按,是用来导出颗粒坐标的,供其它用途。
颗粒提取
颗粒挑选在1k或2k的tomogram仅标注颗粒位置,有了颗粒位置,软件返回到原始tilt series,提取每个颗粒的tilt series,并独立重建每个颗粒。
选择Subtomogram Averaging > Extract particles:
-
勾选
alltomograms -
设置
boxsz_unbin为192。如果在上一步中设置了正确的大小,这里不是必需的,但填了也没什么坏处。 -
输入在挑选颗粒时使用的标签(比如上面说的“initribo”)
-
点击
Launch
等待程序运行完成。
选择Subtomogram Averaging > Build sets:
-
勾选
allparticles -
点击
Launch
这将生成颗粒集合。
-
如果tilt series中存在金颗粒标记,一定要移除它们。参见官网教程。
-
如果从GUI中选择颗粒时box大小就是想要的大小,可以将
boxsz_unbin保留为-1,程序将保持该box大小(自动缩放到原始tilt series) -
如果您的颗粒深埋在其他密度中,使用更大的
padtwod可能会有所帮助,但这样做可能会显著增加内存使用并减慢过程。 -
如果存在CTF信息,可以勾选
wiener,它会在3D重建之前通过SSNR过滤2D颗粒。 -
如果内存/存储/CPU计算资源有限,在
shrink中指定一个缩小因子以生成缩小的颗粒,但这也会限制高分辨率。
初始模型生成
选择Subtomogram Averaging > New initial model generator:
-
设置
particles为刚刚创建的sets/ribo.lst文件(或者刚刚自己取的别的名字) -
可以设置
shrink为2、3或4。越小运行越缓慢,但会生成更好的初始模型,但是实际上目前这一步并不必要 -
增加
batchsize将使用更多CPU核心,并可能在更少的迭代次数中收敛,但每次迭代不会变得更快 -
默认的迭代次数
niter为5,通常完全足够 -
点击
Launch
可以在用文件管理器打开结果(项目管理器右侧的第一个图标)sptsgd_00/output.hdf,看起来不错后在进度监视器(项目管理器右侧的第四个图标)终止程序,已经生成的这个hdf就是后面要用的。
-
如果您的颗粒具有已知的对称性,指定对称性。指定的对称性不会强加到map上,除非还勾选
applysym,不建议在此步骤中勾选此框 -
提取的颗粒的完整采样框大小除以
shrink必须是偶数
模板匹配
用刚刚生成的初始3D模型作为模板,在所有4个tomogram中自动找到所有核糖体。
在此处以及断层扫描重构流程中的任何地方,重建的颗粒具有正对比度(在投影中看起来是白色的),而tomogram和tilt series具有负对比度(在投影中看起来是黑色的)。如果使用来自PDB或其他来源的Reference,应具有正对比度。
选择Subtomogram Averaging > Reference based boxing:
-
Browse选择所有4个tomogram。 -
设置
reference为上一步中生成的output.hdf文件。 -
设置
label为“ribo”,区分于之前手动挑选颗粒的标签 -
设置
nptcl为1000(每个tomogram的最大颗粒数)。如果为了快速测试可以少一点比如设置100,最终分辨率没那么高。 -
vthr默认值10在新版本中有bug,可能会报错,改成5差不多
-
点击
Launch
完成后,可以使用之前使用的Subtomogram Averaging > Manual boxing工具查看所选颗粒,手动删除不良颗粒。
此过程将颗粒位置存储在info文件夹中。
颗粒提取
类似于前面的颗粒提取,只是标签选择上一步模板匹配挑出来的颗粒的”ribo”。
Sub-tomogram Refinement
此步骤使用完整的颗粒集执行常规的迭代sub-tomogram averaging。通常,它将达到15-25 Å的分辨率。由于它涉及多次对完整颗粒集的3D对齐,因此需要大量的计算时间。在下一阶段后将实现更高的分辨率。
选择Subtomogram Averaging > 3D refinement
-
设置
particles为“sets/ribo.lst” -
设置
reference为前面生成的“output.hdf” -
如果怀疑大部分颗粒是坏颗粒,可以尝试减少
pkeep,这将优先排除坏颗粒。 -
设置
threads,根据服务器CPU数量合理设置 -
点击
Launch
结果将逐步保存到spt_01/中。
评估(可选)
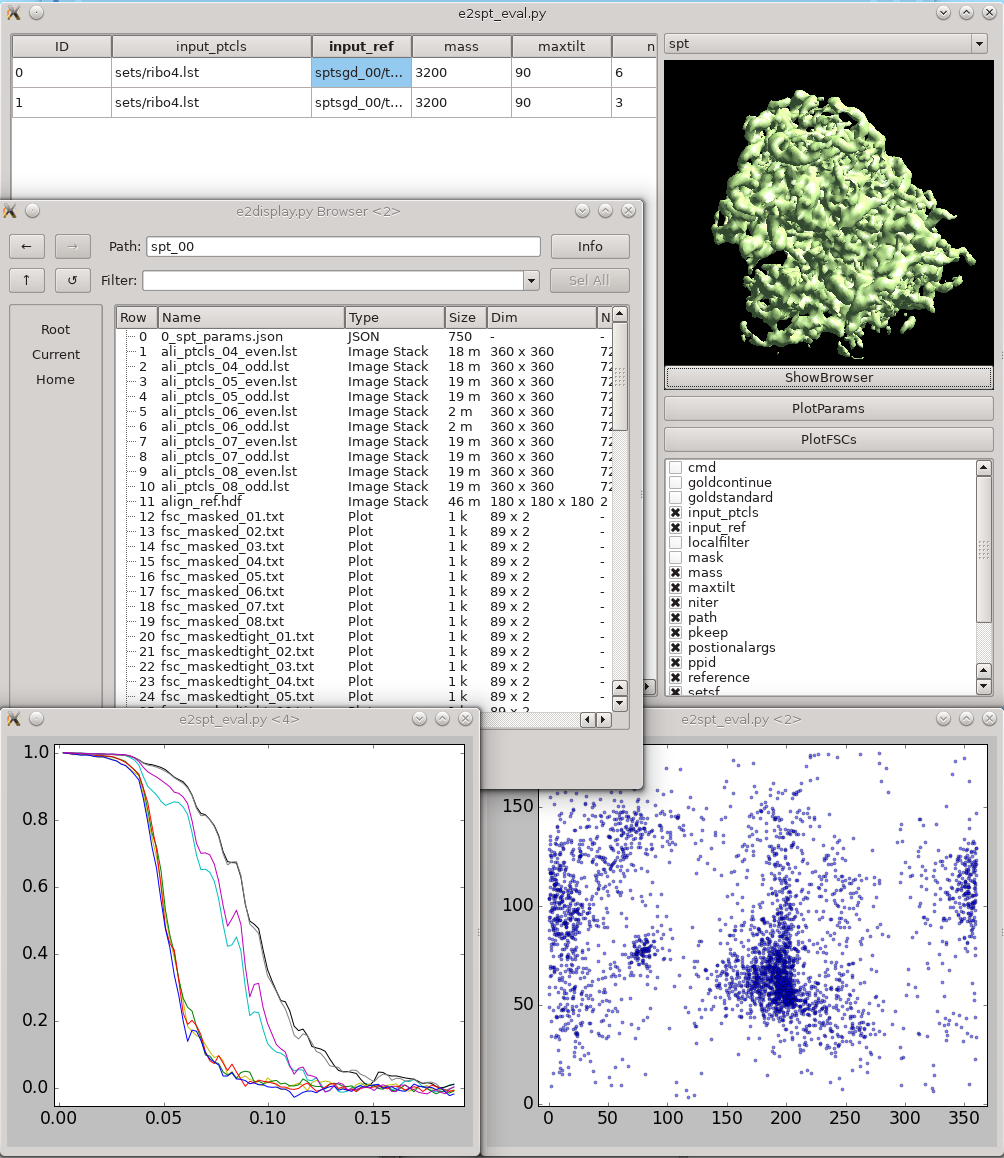
此工具帮助可视化和比较多个sub-tomogram refinement运行的结果。
选择Analysis > Evaluate SPT refinements
基于卷积神经网络的颗粒挑选
点击Subtomogram Averaging > Convnet based auto-boxing,其中label是给挑出来的颗粒的标签,可以自行设置,默认是tomobox。gpuid根据计算机设置就行,也支持没有GPU加速的CNN挑选,但是速度可能会慢个5-10倍。点击Launch。
会弹出四个窗口,一个控制窗口,以及三个颗粒窗口,分别是Positive、Negative和Particle,对应正样本和负样本和预测结果。
在控制窗口中打开一个tomogram,将弹出另外两个窗口:切片视图和颗粒列表。
在切片视图中,左键单击选择样本,Shift+左键单击图像可将其删除,正负样本的控制在控制窗口上面选择Positive还是Negtive,正样本是蓝色的,负样本是红色的。选择少量正样本和负样本,一般十几个就可以。
点击Train开始训练,命令行中会打印迭代过程。继续点击Train或使用更大的Niter值,直到损失值停止下降或者在想要停止的时候停止。损失值并不是绝对的,取决于不同实际数据,所以酌情考虑。
训练完后,点击Apply,让程序使用训练好的网络来选择颗粒。
浏览颗粒列表,Ctrl+左键单击错误识别的颗粒,将其添加到负样本列表中,左键单击颗粒会将其添加到正样本中。也可以再次浏览断层图,将未选中的颗粒添加到正样本中。
可以再次点击Train,使用新的训练集重新训练网络,并点击Apply来检查结果。重复这个过程,直到满意。也可以选择列表中的其他断层图,测试模型的性能,并向训练集中添加更多的正样本和负样本。
浏览列表中的所有断层图,并Apply来选择颗粒。这些颗粒可以在boxer中查看和修改,并通过颗粒提取步骤提取出来。