Phenix,全称Python-based Hierarchical ENvironment for Integrated Xtallography,是一个用于晶体和冷冻电镜结构测定的综合软件包,是辅助结构搭建半自动化的软件之一。使用起来也十分方便,上手很容易,因为大部分过程都是自动的。
Phenix对学术用户是免费的,可以在申请许可后下载。下载后安装也是简单的一路yes。
本文以Phenix v1.20为例,聊聊几个常用的功能。
新建
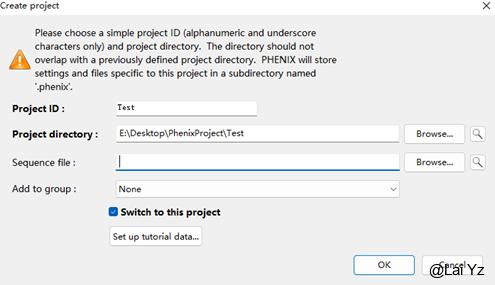
打开Phenix,点击New project,或者第一次打开Phenix,输入名字和保存路径即可。Phenix会在目录下创建一个.phenix的文件夹用于识别以及存储这个project的配置。
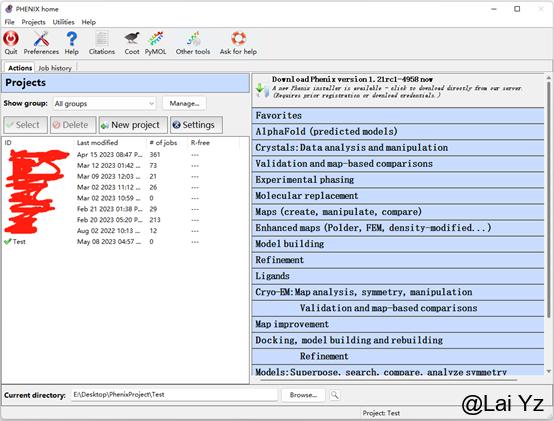
这个就是Phenix的界面,不同的Jobs根据类型分类。Favorites可以存储常用的Jobs。
如果安装过Coot和PyMol,在左上角File-preference-Graphics里可以添加Coot和PyMol的执行程序的路径,这样就可以在Phenix中直接打开这些软件,使用Coot和PyMol可视化结果。
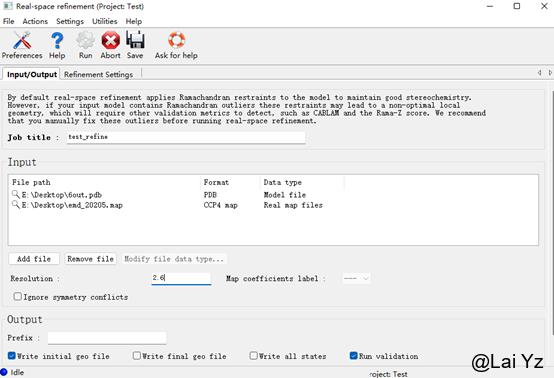
Real Space Refinement
这是用的较多的一个程序,这个程序能基于map对model整体进行优化,包括精细的fit和立体化学校正。

选择add添加map、model,如果model里有不在默认化合物库中的ligand,还需要添加ligand的cif限制文件。cif生成下面eLBOW会说。
这里拿pdb号6out的结构以及对应map EMD-20205为例。
Resolution输入map的分辨率。Job title可写,方便自己识别。
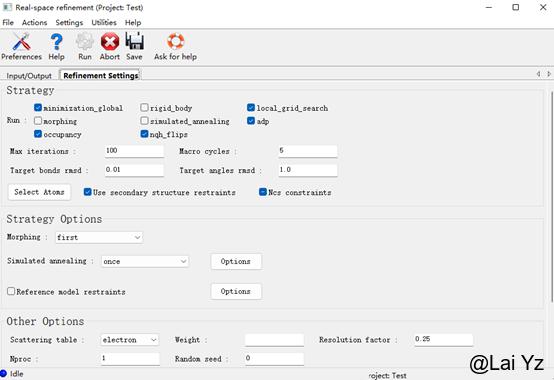
转到Refinement Settings。这里可以调节各种Refine的参数。正常情况下默认的参数就能够有比较好的效果。
由于默认勾选了Use secondary structure restraints,使用二级结构限制,我们需要给出二级结构的范围。如果输入的pdb文件中带了HELIX/SHEET信息,它会自动读取pdb文件中的HELIX/SHEET信息。如果没有,Phenix也有从输入的结构里搜索二级结构的功能。
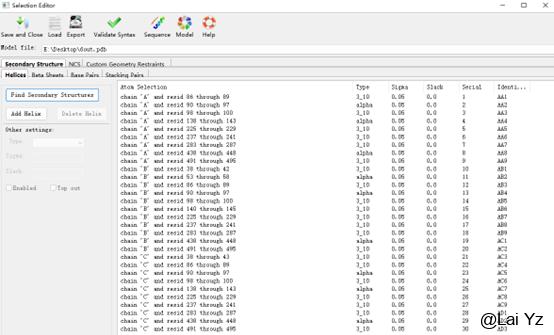
点击Select Atoms。
在Selection Editor中点击Find Secondary Structures,直接点击OK用默认参数搜寻二级结构。然后点击Save and close。
所有参数设置好了,点击Run。输出结果保存在Project目录下,有一个修正好的pdb结构。下方Open in Coot可以直接在Coot中打开。点击结果里的Validation选项,可以看到评估结果。
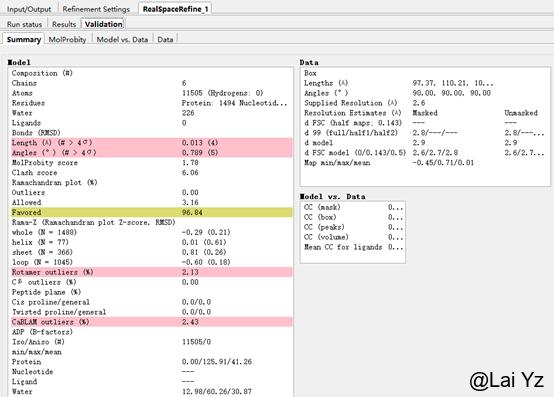
Summary中显示了整体的参数数值,包括Composition (Atoms, Residues, Water, Ligands), Bonds (RMSD), MolProbity score, Clash score, Ramachandran plot, Rotamer outliers, Model vs. Data CC等发表文章时需要在Validation Table中展示的数据。
MolProbity中具体显示了哪些可能有问题的、待修正的氨基酸。包括Clashes、Rotamers、Ramachandran、Bond等Outliers。
大部分情况下,Real space refinement会在restraints的限制下,对model的主链、侧链、键长键角以及立体化学性质进行修正,达到一个较好的水平,但是也存在一些问题、错误和局限性。
① Real space refinement可能会有各种报错,排除软件本身bug,也可能是给的model有问题导致的。
一个可能报错的原因就是缺失cif限制文件,会显示有未识别的原子。看看是不是缺少了哪个model中搭建了的ligand,不在默认的化合物库中,还没有提供相应cif文件。有的时候ligand和cif虽然对应,但是ligand的原子命名和cif的原子命名不匹配也无法识别。比如eLBOW用smile chemical string生成的cif和库里的cif命名就不同。
此外,model最好是标准格式的,前期最好不要添加ligands,如果需要可以使用记事本打开pdb文件删除一些识别不了的条目。
② 对于原本有大范围偏差、偏差较大的位置可能没有很好的修正效果。整体的fit还是在ChimeraX或者Coot里直接手动调整比较合适。
③ 对于一些大侧链比如Trp,有时候不能很好地摆正Flip sidechain。常常会出现Trp的密度很好但是会有侧链翻转180°的现象。这种错误一般都能够在Coot等软件中直观地看到,将其修正过来。
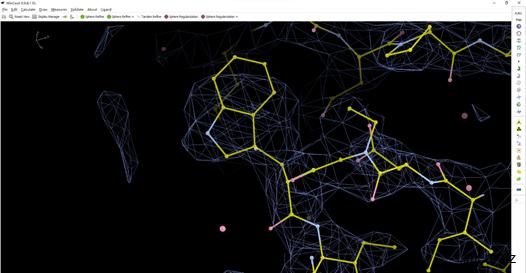
④ 对侧链Rotamer outlier的修正效果在默认参数下有时候会不尽人意。虽然Real space refinement在分辨率较好的情况下能修正大部分的侧链rotamer outlier,但是还是会有一部分的侧链每一次refine都会被调成异常的rotamer。尤其是酸性氨基酸侧链,由于冷冻电子显微镜本身的局限,辐照损伤导致的酸性氨基酸缺失密度,有很多位于外侧的酸性氨基酸密度较差,导致在做完Real space refinement后rotamer outlier很高,后续手动调整就行。
Comprehensive validation (cryo-EM)
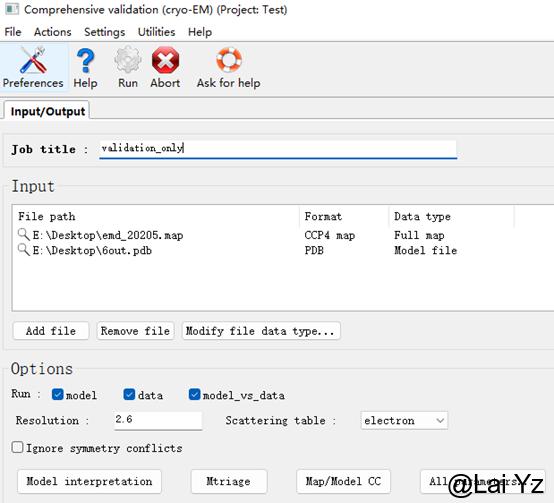
如果只是想做一个Validation,看看有问题的氨基酸或者是生成Validation Reports,而不做refine,那么选择Comprehensive validation (cryo-EM)。

只需要输入model map,有化合物库之外的ligand的输入自定义cif。输入Resolution。点击Run。
结果和Real space refinement里的Validation类似,就不再次说了。
eLBOW
首先要说的是,对于常见的ligand,Phenix和Coot都是由内置cif的库的,直接跑Real space refinement和Validation都是可以的,只有当用一个新的小分子,或者默认的库不合适的时候才来使用这个。因为eLBOW生成的cif有的时候也不是正确的。
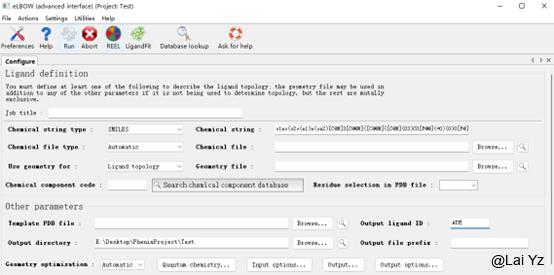
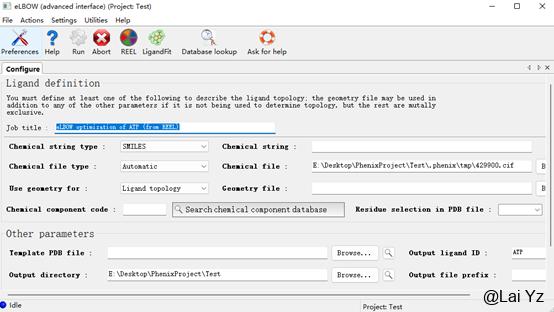
eLBOW (advanced inteface)有更多参数可选,这里就只说advanced了。

打开eLBOW advanced,一般我们可以通过SMILE格式生成cif,这是一种小分子的正则化表示方式,可以在PubChem、RCSB PDB等网站中找到。
以ATP为例,在Chemical string中输入网上搜索到的SMILE。
然后在Output ligand ID中输入三个字母的名字。
点击Run。
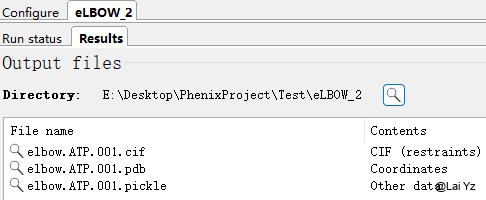
最终会输出ATP的pdb和cif文件。
还有另一种更容易的方法,回到Configure,点击中间的那个Search chemical component database。这个可以在数据库中直接搜索小分子。
输入ATP,点击Search。点击结果中第一行的ATP,然后点击View in REEL。
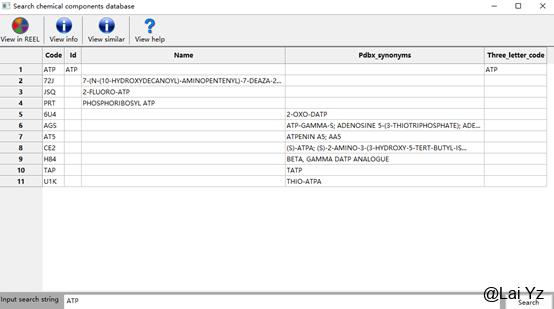
这个就是数据库中的ATP以及键长键角参数。点击右上角的eLBOW Optimisation。
自动打开了一个新的eLBOW advanced界面,由于是从REEL中跳转过来的,直接点击Run就行。
不过一般这种方法能搜到的ligand,默认库中都有。
Combine Focused Maps
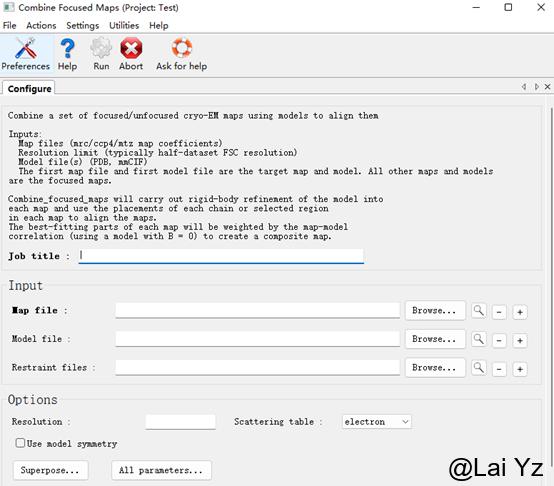
在冷冻电镜数据处理过程中,对于一些柔性较大的区域常常会做Local refine以提高局部分辨率。这个程序可以把不同的Focused maps合并成一个map,保留了Local的高分辨率结构同时又是完整的map。
自带的简介已经写的很清楚了,这个Job需要输入一个主要的map和一个主要的model,通常也就是整体的低分辨map和整体model。然后点击+号可以添加局部的maps和models。
程序会根据model和map进行对齐,匹配得越好的部分会获得更高的权重,最终对map进行合并。
最后会生成一个ccp4格式的map。
其它小工具
这里面有一些修改model的小工具,像PDB Tools PDB file editor可以快速更改修正pdb文件的原子命名、链名、Seg等等参数,Combine PDB可以把两个PDB文件merge。都是一些虽然没啥大用,但是有可能用到并减少工作量的小工具。

除了提到的Jobs,Phenix还有好多工具,都有着各自的作用,许多都可以半自动化,提高结构搭建的效率,减少工作量,可以慢慢摸索。电镜结构搭建时主要经常用的几个程序就是文中提到的这些。
refine自定义限制
在Phenix refine中,Input PDB中所有的LINK,SSBOND,CONECT限制都是自动忽视的,因此想要在refine过程中人为给定义的话就要用特殊的方法。大多数常见的键长键角几何限制都已经在CCP4 monomer库中定义好了,这也是Phenix refine默认调用的parameter。可以通过类似的写法写一个.param后缀的文件来添加额外的键长键角限制。
比如要限制两个His和HEM形成的配位键。新建一个文本文档,根据phenix的语法格式定义键长键角,把文件后缀改成.param。然后在refine程序中选中这个文件,Data Type会自动识别成phil配置文件。
refinement.geometry_restraints.edits {
fe = chain C and resname FE and resid 1000 and name FE
his1 = chain C and resname HIS and resid 99 and name NE2
his2 = chain C and resname HIS and resid 198 and name NE2
bond {
action = *add
atom_selection_1 = $fe
atom_selection_2 = $his1
symmetry_operation = None
distance_ideal = 2.28
sigma = 0.02
slack = None
}
bond {
action = *add
atom_selection_1 = $fe
atom_selection_2 = $his2
symmetry_operation = None
distance_ideal = 2.28
sigma = 0.02 slack = None
}
angle {
action = *add
atom_selection_1 = $his1
atom_selection_2 = $fe
atom_selection_3 = $his2
angle_ideal = 100
sigma = 5
}
}
简单解释一下格式,以refinement.geometry_restraints.edits开头,表明这个一个几何限制文件。然后是原子的选择,可以自己给名字代号,然后根据chain-resname-resid-name分别定义哪条chain的哪个residue name,对应的residueid,以及这个residue中的哪一个原子。这个可以用记事本打开pdb文件找到那个原子的信息。
请问有没有phenix的安装包?如果有的话想求一份
官网直接申请下载应该是免费的。
请问我加了phenix的.param的限制文件,只让小分子和氨基酸成键,但是跑完phenix发现小分子不仅和目标氨基酸成键,还和周围的其他氨基酸有了键,这是为什么呀?
可能强行让它成键以后离附近的氨基酸太近了
您好,我在使用phenix做refine的时候会在点击run之后直接弹出报错,报错内容是动态链接库(DDL)初始化例程失败,请问您有解决措施吗?
没安装好吧,linux装的?
想请教一些Phenix 做real space refine的常用策略是什么呢?(比如推荐使用哪些setting里的工具,refine的迭代次数还有总轮数之类的)
您好,我想请问一下为什么我通过phenix打开coot,显示的就是黑的,没有任何东西,感谢感谢
那还是coot手动打开吧,那个wincoot链接老是有奇怪的bug
有时候我再点一下open in coot就能打开了
老师您好,现在想验证填进去的离子是不是部分占位,但是发现phenix 的refine程序好像是默认按照occupancy1优化,phenix在进行refine时怎么设置修改occupancy的值呀?
老师您好,想问在蛋白核酸复合物中,如何计算蛋白和核酸分别的b-factor值
phenix的validation summary里有写
老师好,请问Combine Focused Maps这个功能是不是running比较慢,导入的map是用sharp的map吗?
看map大小和你电脑的水平吧
sharp的