蛋白质设计基本的流程是:生成蛋白质骨架,生成骨架氨基酸序列,结构预测验证折叠以及相互作用,实验验证。
本文涉及的源代码都已经开源,在Github中能找到和以及。更多参数的更改请自行查阅github仓库指南或论文。
RFdiffusion
1. 安装
首先是RFdiffusion的安装
先克隆源代码
git clone https://github.com/RosettaCommons/RFdiffusion.git
然后可以下载模型权重,需要时间比较久,一般使用的话下载下面前几个模型就行,最后两个可以不下载。
cd RFdiffusion
mkdir models && cd models
wget http://files.ipd.uw.edu/pub/RFdiffusion/6f5902ac237024bdd0c176cb93063dc4/Base_ckpt.pt
wget http://files.ipd.uw.edu/pub/RFdiffusion/e29311f6f1bf1af907f9ef9f44b8328b/Complex_base_ckpt.pt
wget http://files.ipd.uw.edu/pub/RFdiffusion/60f09a193fb5e5ccdc4980417708dbab/Complex_Fold_base_ckpt.pt
wget http://files.ipd.uw.edu/pub/RFdiffusion/74f51cfb8b440f50d70878e05361d8f0/InpaintSeq_ckpt.pt
wget http://files.ipd.uw.edu/pub/RFdiffusion/76d00716416567174cdb7ca96e208296/InpaintSeq_Fold_ckpt.pt
wget http://files.ipd.uw.edu/pub/RFdiffusion/5532d2e1f3a4738decd58b19d633b3c3/ActiveSite_ckpt.pt
wget http://files.ipd.uw.edu/pub/RFdiffusion/12fc204edeae5b57713c5ad7dcb97d39/Base_epoch8_ckpt.pt
# 可选下载的beta模型:
wget http://files.ipd.uw.edu/pub/RFdiffusion/f572d396fae9206628714fb2ce00f72e/Complex_beta_ckpt.pt
# 原始结构预测模型:
wget http://files.ipd.uw.edu/pub/RFdiffusion/1befcb9b28e2f778f53d47f18b7597fa/RF_structure_prediction_weights.pt
然后创建conda环境。确认在RFdiffusion目录下了,然后:
conda env create -f env/SE3nv.yml
conda activate SE3nv
cd env/SE3Transformer
pip install --no-cache-dir -r requirements.txt
python setup.py install
cd ../.. # 进入RFdiffusion的根目录
pip install -e .
我在我的服务器上用这个默认的SE3nv.yml环境不成功,所以自己改了一套环境。可以根据自己的显卡及电脑配置尝试一下,安装好后跑一个测试脚本看是否通过。一般都是cuda、pytorch之类的版本依赖问题。下面是我的的yml,如果自动安装的numpy版本太高到了numpy2,使用时报错,就降级到numpu1就行(比如numpy-1.26)
name: SE3nv
channels:
- pytorch
- nvidia
- defaults
- conda-forge
- dglteam
dependencies:
- python=3.9
- pytorch=1.13.1
- pytorch-cuda=11.7
- torchaudio=0.13.1
- torchvision=0.14.1
- cudatoolkit=11.7.1
- dgl-cuda11.7
- pip
- pip:
- hydra-core
- pyrsistent
再次使用只要用conda activate SE3nv激活环境就行。
2. 基本使用
假设RFdiffusion安装位置为/opt/RFdiffusion。测试输出的位置在/path/to/testdir/。
RFdiffusion主要运行脚本为/opt/RFdiffusion/scripts/run_inference.py
最基础的测试:
/opt/RFdiffusion/scripts/run_inference.py 'contigmap.contigs=[50-80]' inference.output_prefix=/path/to/testdir/test inference.num_designs=3
这是一个最基础的预测命令,实现的是随机生成一个氨基酸长度为50到80个的多肽(即固定50)
contigmap.contigs是指定序列的参数,如果纯数字,前方没有字母,就代表着生成序列。保存目录是/path/to/testdir/,生成的多肽结构文件名前缀是test,总共生成3个结构。
有许多命令的脚本示例都放在/opt/RFdiffusion/examples在中,比如这条基本的生成命令,可以直接通过运行/opt/RFdiffusion/examples/design_unconditional.sh,用来检测环境是否安装成功。
3. Binder Design
这是一个针对某个target设计binder基本的命令:
/opt/RFdiffusion/scripts/run_inference.py \
'contigmap.contigs=[B1-100/0 100-100]' \
inference.output_prefix=/path/to/testdir/binder_test \
inference.input_pdb=/path/to/testdir/target.pdb \
inference.num_designs=10
这个命令会对输入结构的B链1-100氨基酸设计一个binder。
主要是添加了一个inference.input_pdb来指定输入的pdb,以及contigmap.contigs的区别。B1-100代表了B链的1-100位氨基酸作为target,/0是添加链分隔,然后100-100是生成100个氨基酸的binder。
设计所耗费的时间取决于蛋白的大小,所以最好在预测前修剪一下结构。但是这导致了一个问题,修剪结构可能会暴露蛋白内部的疏水核心,从而使binder很容易设计在这些不可能结合到的位置。解决的办法是添加hotspot,这样程序就会在这些hotspot附近去设计binder。例如添加参数:
'ppi.hotspot_res=[A30,A33,A34]'
就可以在A链的30、33、34位氨基酸附近随机设计binder。不是所有的hotspot都会同时使用,官方建议给3-6个hotspot,并在大规模设计前测试一下哪些hotspot好用。
根据官方的说法,对于不同难度的target,设计1000-10000个binder骨架,每个生成1-2个氨基酸序列,通常足以获得比较好的binder。
4. Motif Scaffolding
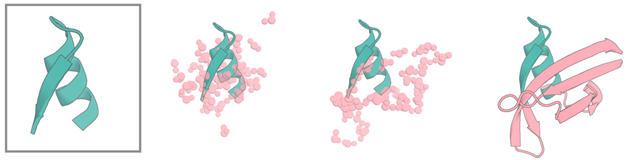
RFdiffusion可以针对某段motif再设计。如果我们有了一个target和binder结合的结构,想要保留这段motif,保留结合位点的同时寻找更多稳定多样甚至结合更好的binder,就可以用Motif Scaffolding。
/opt/RFdiffusion/scripts/run_inference.py \
inference.output_prefix=/path/to/testdir/design_motifscaffolding \
inference.input_pdb=/path/to/testdir/input.pdb \
'contigmap.contigs=[5-15/A10-25/30-40/0 B1-100]' \
inference.num_designs=10
这里contigmap.contigs参数改成了[5-15/A10-25/30-40/0 B1-100]
它的意思是保留A链(作为binder)的10-25位氨基酸,在N端加入随机5-15个氨基酸,在C端加入随机30-40个氨基酸。/0代表了链的分隔。target是B链的1-100位氨基酸。
或者,反过来也是一样的,可以通过inpaint_seq参数来覆盖掉某些氨基酸
比如添加参数'contigmap.inpaint_seq=[A1/A30-40]',这样,A链的1号和30-40号氨基酸就会被遮盖掉,从而重新设计。
另外,如果想要在两端都随机加入氨基酸的同时,保持设计链的总长度为55个氨基酸的话,再加入一个参数就可以:contigmap.length=55-55。这个没有单引号。
如果要设计的结合motif非常小,默认的RFdiffusion模型可能效果会差一些,可以用微调过的模型,加入参数:inference.ckpt_override_path=/opt/RFdiffusion/models/ActiveSite_ckpt.pt。如果要调节使用别的模型权重也都是使用这个参数inference.ckpt_override_path=
5. Partial diffusion
我们可以用RFdiffusion来对结构进行部分添加噪音和去噪,来对一个蛋白重新设计,保留一定的基础结构。

比如使用这两个参数:'contigmap.contigs=[100-100/0 B1-150]' diffuser.partial_T=10
target链是B1-150,同时对A链进行重新设计,diffuser.T越大会增加binder的多样性,默认的diffuser.T是50。
这种情况下输入的A链必须刚好是100个氨基酸,不然程序会不知道多余的氨基酸从哪里延申。
另外,对于diffuse的部分,如果想要固定某段序列,可以这样:
'contigmap.contigs=[100-100/0 20-20]' 'contigmap.provide_seq=[100-119]' diffuser.partial_T=10
这样这20个氨基酸就固定了。provide_seq会使用不同的模型,但是这个程序会自动识别并选择。
6. Symmetric Oligomers
RFdiffusion还可以设计对称的多聚体,目前支持循环对称cyclic、二面体对称dihedral和四面体对称性tetrahedral。它需要用到另一个config文件来进入symmetry模式。
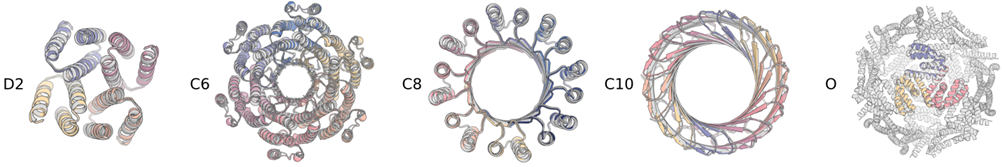
/opt/RFdiffusion/scripts/run_inference.py \
--config-name symmetry \
inference.symmetry=tetrahedral \
'contigmap.contigs=[360]' \
inference.output_prefix=/path/to/testdir/test_tetrahedral \
inference.num_designs=1
这条命令会生成一个360个氨基酸的四面体对称性蛋白。inference.symmetry也可以是c2或者d4这种形式。contigmap.contigs代表了总氨基酸数量,所以必须能除尽对称数。
对称性多聚体同样可以用Motif Scaffolding,基于某些motif来生成蛋白。例如:
/opt/RFdiffusion/scripts/run_inference.py \
inference.input_pdb=/opt/RFdiffusion/input_pdbs/nickel_symmetric_motif.pdb \
'contigmap.contigs=[50/A2-4/50/0 50/A7-9/50/0 50/A12-14/50/0 50/A17-19/50/0]' \
inference.symmetry="C4" \
inference.num_designs=5 \
inference.output_prefix=/path/to/testdir/design_nickel
Symmetric Motif Scaffolding需要先提供一个有对应对称性的pdb,在${base_dir}/input_pdbs中有一个例子是一个由四个His配位一个Ni离子的核心pdb。
contigmap.contigs在每个His两端加入50个随机氨基酸,总共四条链。inference.symmetry给C4对称性。
这样就可以设计出一个四聚体,保留初始给的四组氨酸核心。
如果去看examples里的对应脚本会看到还额外设置了potentials,并用了另外一个权重模型。可以通过使用Auxiliary Potentials来干预去噪过程来指导diffusion过程。不同的权重效果不同,需要实际测试来评估和选择。详情可以见Github仓库和文章。
7. Fold Condition
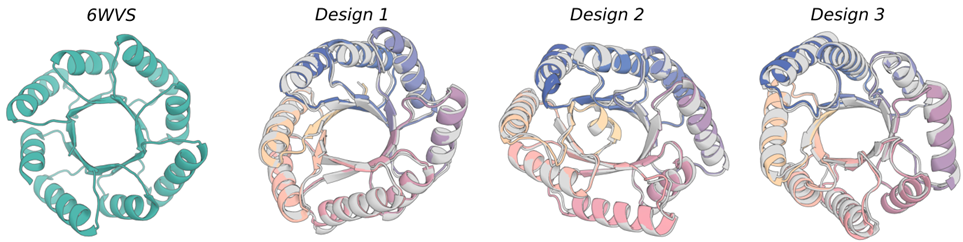
有的时候在特定拓扑上结构上优化蛋白质设计效果很好,通过给特定模型额外提供二级结构和块邻接信息实现的。
首先我们需要生成二级结构(ss.pt)和块邻接信息(adj.pt)文件。用到helper_script中的脚本。
# 针对一个pdb
/opt/RFdiffusion/helper_scripts/make_secstruc_adj.py --input_pdb /path/to/testdir/input.pdb --out_dir /path/to/test
# 针对多个pdb
/opt/RFdiffusion/helper_scripts/make_secstruc_adj.py --pdb_dir /path/to/testdir/pdbs/ --out_dir /path/to/test
这样可以为现成的拓扑结构的蛋白质生成pt文件,然后可以用来当模板。
/opt/RFdiffusion/scripts/run_inference.py \
inference.output_prefix=/path/to/testdir/testfc \
scaffoldguided.scaffoldguided=True \
scaffoldguided.target_pdb=False \
scaffoldguided.scaffold_dir=${base_dir}/examples/ppi_scaffolds_subset
这个${base_dir}/examples/ppi_scaffolds_subset里有一个提供的库,大部分是一些现成的α螺旋肽,也可以自己另外提供。
此外,如果做PPI的话,官方还推荐将input pdb也做一个ss.pt和adj.pt提供。
/opt/RFdiffusion/scripts/run_inference.py \
scaffoldguided.target_path=/path/to/testdir/input.pdb \
inference.output_prefix=/path/to/testdir/ppi_scaffolds_test \
scaffoldguided.scaffoldguided=True \
'ppi.hotspot_res=[A59,A83,A91]' \
scaffoldguided.target_pdb=True \
scaffoldguided.target_ss=/path/to/testdir/input_ss.pt \
scaffoldguided.target_adj=/path/to/testdir/input_adj.pt \
scaffoldguided.scaffold_dir=/opt/RFdiffusion/examples/ppi_scaffolds/ \
inference.num_designs=10 \
denoiser.noise_scale_ca=0.5 \
denoiser.noise_scale_frame=0.5
这样一条命令首先需要对input.pdb先生成pt,使用官方提供的ppi_scaffolds库,然后靶向hotspot生成蛋白质骨架。denoiser.noise_scale噪声多少折中一下多样性和设计蛋白的可行性。
dl_binder_design
安装
这个仓库分为两个部分,ProteinMPNN-FastRelax和AlphaFold2,最好分开conda环境管理,能减少一些依赖错误,如果有能力放到一个环境也行。 另外,这两个都需要PyRossetta,在conda channel中添加许可证。这个自己去pyrosetta申请或者设置。
然后这个仓库不包含ProteinMPNN的源代码,所以需要下载至${base_dir}/mpnn_fr。
另外,涉及到AF2,还需要下载alphafold2的模型权重。
git clone https://github.com/nrbennet/dl_binder_design.git
cd dl_binder_design/mpnn_fr
git clone https://github.com/dauparas/ProteinMPNN.git
cd ../af2_initial_guess
mkdir -p model_weights/params
cd model_weights/params
wget https://storage.googleapis.com/alphafold/alphafold_params_2022-12-06.tar
tar --extract --verbose --file=alphafold_params_2022-12-06.tar
首先安装ProteinMPNN-FastRelax的环境,进入dl_binder_design/include目录
conda env create -f proteinmpnn_fastrelax.yml
我装的时候遇到了一些依赖错误,降级一下mkl=2024.0 numpy=1.26就能运行。
作者提供了一个导入测试来判断基本的安装是否成功,装好之后激活环境,然后运行
python /opt/dl_binder_design/include/importtests/proteinmpnn_importtest.py
如果一切正常,就通过了基本测试。
AlphaFold2的安装依赖问题比较复杂,所以作者给了适配两种环境的yml文件af2_binder_design.yml(cuda12)和dl_binder_design.yml(cuda11.1)。需要另外cuda版本的自行修改yml文件。
conda env create -f af2_binder_design.yml
我在使用af2_binder_design.yml时还需要将biopython版本降至1.81,scipy版本降至1.12.0。同样用导入测试来验证安装结果。
python /opt/dl_binder_design/include/importtests/af2_importtest.py
在/opt/dl_binder_design/include/silent_tools中含有silent文件格式转换工具,需要将这个目录添加到环境变量中方便使用。
silent文件
官方建议使用silent文件,比起上千上万个pdb结果,将他们打包在一个silent文件里有时候更方便一些。
将pdb合并成单个silent文件
silentfrompdbs *.pdb > my_designs.silent
将silent文件解压成pdb
silentextract all_structs.silent
另外的silent命令可以自己再去看。
如果需要固定氨基酸,不让MPNN重新设计这段区域,可以在helper_script中找到对应的脚本。
python /opt/dl_binder_design/helper_scripts/addFIXEDlabels.py --pdbdir /dir/of/pdbs --trbdir /dir/of/trbs --verbose
这样自动将RFdiffusion生成的骨架的固定区域添加FIXED标签,再将这些pdb打包成silent进行后续操作就可以了。
ProteinMPNN-FastRelax
使用ProteinMPNN-FastRelax很简单:
/opt/dl_binder_design/mpnn_fr/dl_interface_design.py -silent /path/to/my_designs.silent -outsilent /path/to/my_designs_mpnn.silent
-outsilent不是必须的,如果没有指定保存的名字,会自动保存out.silent。
如果不用silent,用的pdb文件,就用-pdbdir
/opt/dl_binder_design/mpnn_fr/dl_interface_design.py -pdbdir /path/to/backbone_pdbs/ -outpdbdir /path/to/outpdbs/
AlphaFold2 intial guess 生成的结果用AlphaFold2进行预测评估。
/opt/dl_binder_design/af2_initial_guess/predict.py -silent /path/to/my_designs_mpnn.silent -outsilent /path/to/out.silent -scorefilename /path/to/out.sc
outsilent和scorefilename 都不是必须的,如果不指定,会自动保存out.silent和out.sc。out.silent就是经过AlphaFold2 initial guess预测的结构结果,out.sc就是score文件,包含了pae分数和pldtt分数等,用于筛选合适的预测。官网给普通binder建议pae_interaction小于10用于实验验证。还需要结合自己的需求来筛选判断
不用silent用pdb也是类似。
/opt/dl_binder_design/af2_initial_guess/predict.py -pdbdir /path/to/pdbs/ -outpdbdir /path/to/af2out/ -scorefilename /path/to/out.sc
BindCraft
BindCraft是一个更加全自动的binder设计程序,它生成骨架的方法是基于AF2,据说成功率十分高。
安装
安装也很自动化,官方提供了一个脚本:
git clone https://github.com/martinpacesa/BindCraft
cd BindCraft
bash install_bindcraft.sh --cuda '12.4' --pkg_manager 'conda'
使用
BindCraft输入参数是读取json参数文件。基础的命令格式就是:
python -u /opt/BindCraft/bindcraft.py --settings 'target.json' --filters 'filters.json' --advanced 'advanced.json'
这几个参数文件的基础模板可以分别在/opt/BindCraft下的settings_target/PDL1.json, settings_filters/default_filters.json, settings_advanced/default_4stage_multimer.json 下找到并查看。后两者是调节权重阈值参数的,主要是target.json每次需要修改:
{
"design_path": "/content/drive/My Drive/BindCraft/PDL1/",
"binder_name": "PDL1",
"starting_pdb": "/content/bindcraft/example/PDL1.pdb",
"chains": "A",
"target_hotspot_residues": "56",
"lengths": [65, 150],
"number_of_final_designs": 100
}
这些参数基本都能看出是干嘛的,复制一份然后都改成自己设计需求对应的文件和参数就行。
运行后它就能自动进行设计,并根据阈值参数进行过滤,直到生成足够多通过阈值的目标。
ProteinMPNN-FastRelax的输入文件必须包含binder和蛋白质文件吗,我只用蛋白质文件会报错
应该是给binder文件就行
那在af2_binder_design输入文件中,是只要binder文件就行吗,那它是怎么预测binder和蛋白的相互作用的
可以请问一下,怎么申请pyrosetta的学术账号吗?