
ISOLDE是一个新兴的结构搭建软件,尤其是针对中低分辨率结构,可以在分辨率不佳的位置尽可能的搭建合理的氨基酸结构,减少Clashes,但是立体化学的权重稍微低一些。并且相对于Coot,它的自动化程度更高,更易上手。
ISOLDE是基于分子动力学模拟的。它借助了许多大名鼎鼎的工具包。利用AMBER和CHARMM等的forcefields文件对分子提供了高保真的力学特性描述。使用OpenMM、AMBER等的分子动力学引擎,利用GPU强大的大规模并行运算能力,能够求解几千个原子每秒数百次的运动。使用了ChimeraX提供了一个良好的交互式界面。
由于ISOLDE是作为ChimeraX的插件安装的,因此需要ChimeraX的基础。建议先学习ChimeraX的教程。
本文的教程基于与官网Tutorial,使用的是诺如病毒衣壳蛋白VP1的结构。
ISOLDE也有,想要系统学习或者遇到问题时可以查阅。
ISOLDE的安装
ISOLDE最好在有GPU(最好是Nvidia显卡,GTX 1060以上,显存越大越好)的情况下使用,会有更流畅的体验。
首先下载ChimeraX。
ISOLDE版本是随着ChimeraX版本更新的,需要先,至少下载ChimeraX v1.3以上才有ISOLDE。目前已经是v1.8。
然后打开ChimeraX,点击工具栏的Tools-More Tools…
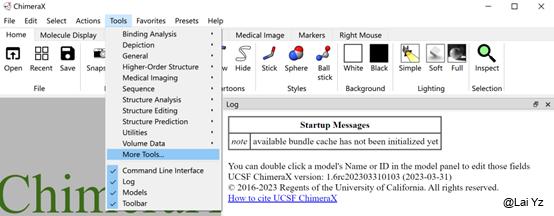
往下拉在Top Downloaded Bundles里就能看到ISOLDE。
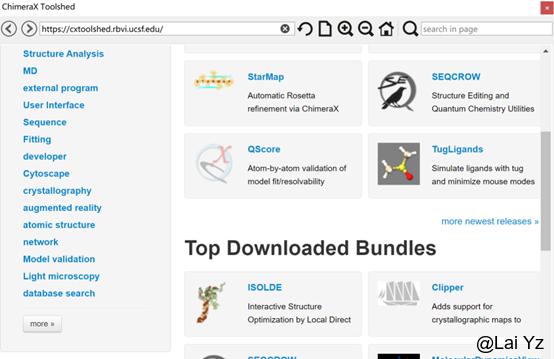
(本人的尝试中,这里需要翻墙,不然Install的图标加载不出来,甚至校园网连More Tools…都加载不出来。如果没有梯子,往下看有方法。)
点击蓝色的Install按钮下载安装包。
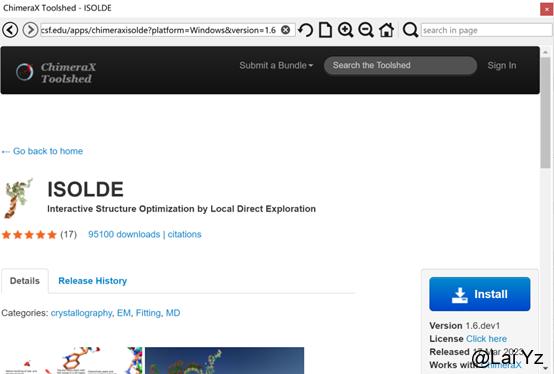
下载好后,会弹出Toolshed对话框,如果翻墙下载的朋友在这里点击Toolshed - Install之前记得关闭梯子,不然会报错。
如果弹出这个对话框代表安装成功了,重启ChimeraX。
此时工具栏已经添加了ISOLDE。
如果没有梯子,可以找有的人要过来ChimeraX_ISOLDE-1.8-cp311-cp311-win_amd64.whl文件,用Toolshed命令也能安装。这是v1.8的,需要配套ChimeraX v1.8
下载好了以后在ChimeraX的下方命令行里输入toolshed install your/path/to/ChimeraX_ISOLDE-1.8-cp311-cp311-win_amd64.whl。记得把path改成你存放whl文件的位置。
启动ISOLDE
打开Tutorial数据,是pdb 6out的一个历史版本的结构。在命令行输入:
isolde demo cryo_em_intro modelOnly true startIsolde false
再从EMDB数据库下载对应的map。在命令行输入:
open 20205 from emdb
我们在上方工具栏找到ISOLDE选项,点击Start ISOLDE。
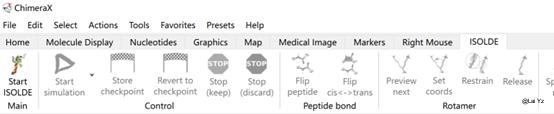
ISOLDE启动后会显示这个框。这就是ISOLDE的主界面。
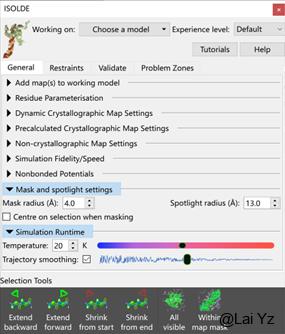
首先我们需要选择working model和map。
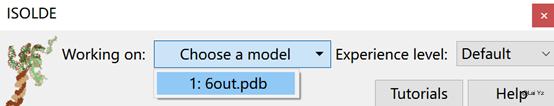
点击choose a model选6out。
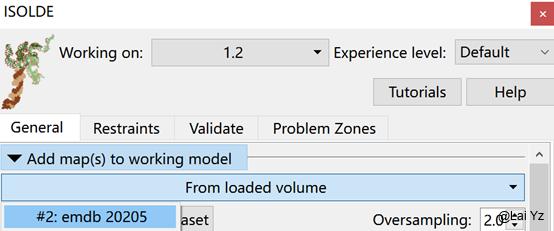
然后点击Add map to working model选择emdb 20205。
基本操作和显示
1、控制
ISOLDE的基本控制和ChimeraX有一点点不一样。
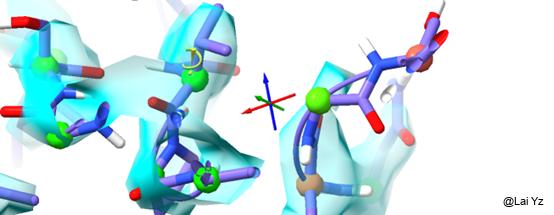
红蓝绿的十字代表中心位置。
鼠标左键:旋转。
鼠标右键:移动。
鼠标中键:移动。
鼠标滚轮:放大/缩小。
Alt+鼠标滚轮:调节Contour level。
2、主链Cα碳原子用球表示,根据Ramachandran plot会显示不同颜色。颜色由绿色到橙色到红色表示越来越异常的构象。
3、可能的Rotamer outlier用螺旋加感叹号的符号显示。颜色由橙色到红色表示越来越异常的构象。
4、Clash无法像Coot一样显示在屏幕上。ISOLDE用了分子动力学模拟,在refine的时候自动计算能量低的位置,减少主链侧链构象的权重,同时减少clash。也正是因为这样,如果结构本身有严重的clash,比如原子几乎重叠,那么ISOLDE将无法运算,因为能量是无穷大,根据模拟原子会直接飞到无穷远。
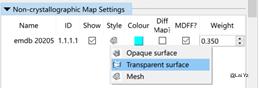
5、Map的显示的话我个人不太喜欢mesh,更喜欢transparent surface,可以在General-Noncrystallographic Map Settings里调。
这里还可以调节MDFF(Molecular Dynamics Flexible Fitting),这个是默认打开的,我们可以调节权重,类似于Coot里的refine weight。
前处理
把背景调成白色。
1、加氢
ISOLDE需要含氢原子的model进行分子动力学模拟。如果model不含氢原子,可以在底部命令行Command输入addh来加氢。
2、限制轻分子
此外,因为这个结构里有水分子,我们还需要对水分子加以限制。
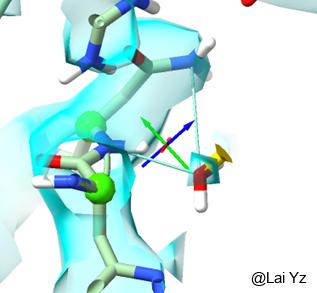
isolde restrain ligands #1
这条命令会自动把含有三个及以下重原子的ligands和附近的原子绑定,不至于在模拟的时候因为ligands太轻、密度强度相对较弱而飞出去。如果附近没有相互作用原子,那么它会绑在原地。这个绑定是很弱的,仅仅防止它飞出去,允许正常的运算。如图所示黄色图钉图标。
在Restraints选项里也可以对任何原子手动添加restraints,在别的一些情况下用得到。没有需要的话限制这一步可以跳过。
Validation
点击ISOLDE里validate选项卡,有如下几个板块。在这里我们可以看到结构中可能存在异常的氨基酸或分子。
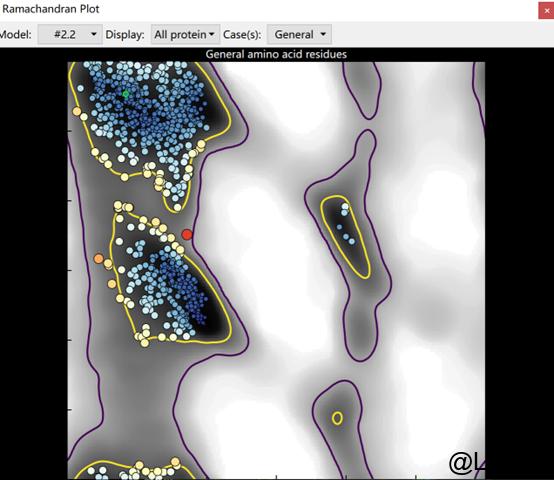
点开Validate-Ramachandran Plot,展示拉式图。Disallowed的氨基酸会标记成红色的圆点,我们点击它就可以在移到对应的位置上去。
Peptide Bond Validation Rotamer Clashes分别以表格形式展示显著的Bond outlier、Rotamer outlier和Clashes。
Unparameterised Residue展示的是ISOLDE无法识别的单位。一般是没有正确加氢或者缺原子的氨基酸,以及一些不匹配库的ligands。
如果存在Unparameterised Residue,运行的时候会报错并让确认。它会展示库中与当前分子最像的Residue,点一下把对应的单位修正就行。
虽然ISOLDE包含了上万个ligands库,且在不断更新中,还是有一些ligands不在库里,那就只能自己提供限制文件。
ISOLDE和Coot不一样,不使用cif来限制,而使用OpenMM的XML格式的forcefield文件来描述ligands的分子动力学参数,比较复杂。
Simulation
Simulation就类似于Coot里的refine,ISOLDE会对选中的原子及附近的氨基酸进行分子动力学模拟。也可以选中整个结构进行模拟,来修正许多容易修正的错误。前提是结构不要太大,得在机器的运算能力之内,否则容易卡死。
ISOLDE也允许牺牲模拟精度来提升速度。在General-Simulation Fidelity/Speed里,默认是最高精度-最低速度。不建议降低精度。
选中全部原子,在command line输入:
select #1
点击ISOLDE工具栏Start simulation。可以看到原子开始振动,进行分子动力学模拟。
这里我的电脑是NVIDIA RTX3060的显卡,整体的simulation还是比较卡,局部的明显流畅许多。
点击左上角Store checkpoint会存储当前状态。Revert to checkpoint会撤回到上一个checkpoint。
注:每次点击Start simulation会先自动存储一次当前状态。
Stop(keep)会停止模拟并保留修改。
Stop(discard)会停止模拟并不保留修改。
对氨基酸的修正
正常情况下就是在Validate里或者自己寻找搭建有问题的氨基酸,然后用Simulation修正。
Simulation一般会自动进行修正,同时,我们也可以自行调整。
在Peptide Bond里,Flip peptide选项可以翻转选中氨基酸的肽平面,Flip cis<->trans可以在顺(cis)反(trans)构象之间转换。
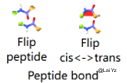
在Rotamer里,Preview next可以显示选中氨基酸所有可能的最优构象,Set coords则可以将其应用。
还有一个功能是Problem Zone。ISOLDE自动分析了存在问题比较多的区域,可能有较多的错误,可以一次修正完。
添加Ligands
Command输入isolde add ligand可以添加ISOLDE库中包含的ligands,后面需要跟三个字母的ligand命名。
比如
isolde add ligand SF4
就生成了一个四铁四硫簇。