Coot是经典的晶体及电镜结构可视化搭建软件,可以从官网直接下载对应版本:
链接:
也可以在CCP4官网下载CCP4软件包,包含了Coot:
推荐是直接安装全家桶CCP4软件包,只安装Coot会缺失一些功能比如MolProbity、Clash、Ligand等,需要另外安装这些拓展,比较麻烦。
这些都是免费的,并且支持Windows、MacOS、Linux操作系统。

Coot有官方的Manual文档十分详细,有不懂的功能可以查询:
本文使用的是发表在PNAS上的人琥珀酸脱氢酶的结构,EMDB号为。起始同源模型使用的是孙飞老师和饶子和老师2005年发表在Cell上的猪琥珀酸脱氢酶,PDB号为。
基本界面和操作
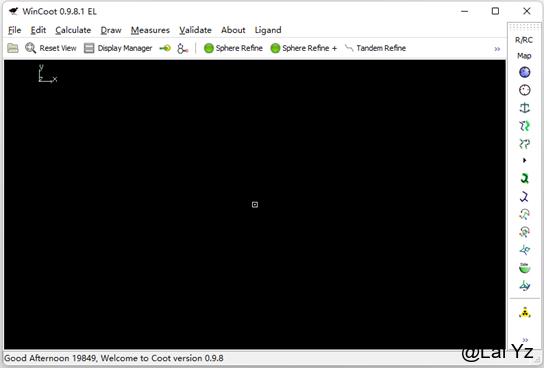
打开Windows版本的Coot后是这样的界面。右侧有一个可以自定义的工具栏,包括了许多常用的工具。
通常还会弹出一个命令窗口,可以方便查看进程和报错。
File - Open Map.. 选择EMD-34255。注意文件的路径尽量不能有中文,有些情况可能会打不开文件。
Edit - Map Parameter - Map Radius EM 可以调节Map显示的大小,越大当然可能会越卡,根据电脑的性能酌情调节。这个Map调到70就基本覆盖了当前这个整个模型了。我的电脑一般调到30会比较流畅。
几个常用的鼠标基本操作:
-
鼠标左键拖动可以旋转
-
鼠标右键拖动可以放大缩小
-
Ctrl+鼠标右键拖动可以调节显示的远近
-
鼠标滚轮滚动调节Contour level
-
鼠标滚轮拖动或者Ctrl+鼠标左键拖动可以移动视角
-
鼠标双击显示/取消原子信息标记
-
空格/Shift+空格 前进/后退一个氨基酸
Roughly Fit
这里我们用到了一个同源模型-猪的琥珀酸脱氢酶结构,以往我们在自己搭建一个新的模型的时候一般是用同源模型作为初始结构搭建的,然后将氨基酸突变成自己的蛋白。
现在有了许多蛋白质结构预测软件,又给了我们另一个选择。我们现在搭建模型的时候会用AlphaFold预测的模型Fit进去然后修正,省去了同源模型突变的过程,减少了很多工作量。
还有一个选择是用model_angelo这类自动建模软件,如果分辨率足够高的话,给它map和序列就能生成一个完全fit的model,然后再手动精修。
总而言之,现在技术发展的很快,选择有很多,减少了我们很多的工作量,这里讲一下传统的方法,在其他方法不合适的时候能用上。
从网上下载下来的同源模型的的位置一般和密度的位置不在一起,需要先粗略地将其摆进密度里。这个可以用Coot的Morph-Jiggle Fit,或者Phenix、Chimera都有类似的Fit、Dock这类的功能。
这里用ChimeraX将1ZOY结构Fit进EMD-34255里。然后导出一个新的位置正确的pdb。
突变氨基酸(提取部分结构)
File - Open Coordinates… 选择刚刚Fit过保存的1zoy_fit.pdb打开原子模型。
在Display Manager里可以将Molecular显示方式改成C-alphas/Backbone。这样会只显示主链碳原子,在调整整体时会清爽一些。选择CAs+Ligands可以显示主链碳原子的同时显示配体。其它不同的显示方式可以自己尝试一下看看。快捷键是Ctrl+Shift+滚轮在不同方式中切换。
上方工具栏的Draw > Sequence View 可以展示氨基酸序列。

为了方便,我们这里先提取一条链Chain B教学。
Edit > Copy Fragment
Atom selection for fragment 输入//B/9-247
点击OK
然后打开Display Manager可以看到生成了一个新的分子叫atom selection from 1zoy_fit.pdb。我们可以把原来的直接删掉或者不显示。
现在应该就是这个样子。
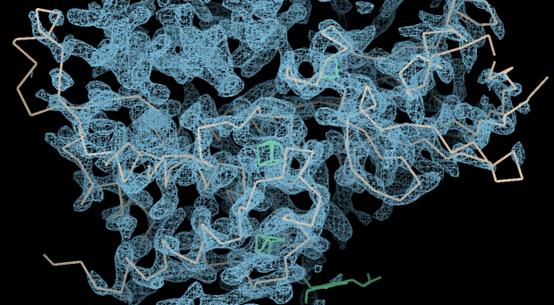
然后我们将猪的氨基酸序列突变成人的。
Calculate > Use Clustalw for alignment, then mutate 分子选当前分子,Chain-ID选择B,Sequence file在Uniprot上下载人对应的序列。

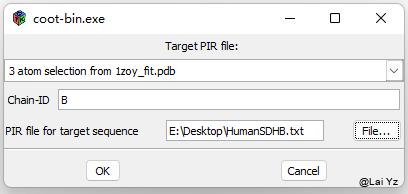
或者用另一种半自动的方法。
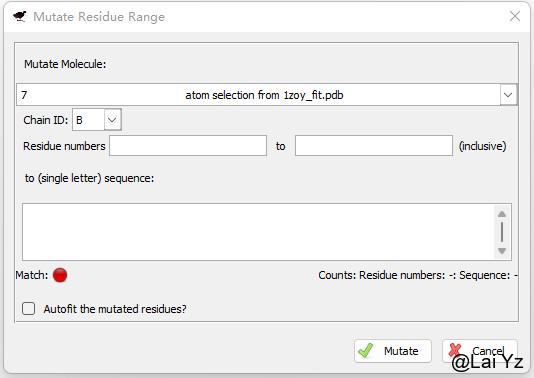
Calculate > Mutate Residue Range 确定Residue numbers的范围,在下方输入要突变成的序列。
这个方式更加明确一些,但是需要确保numbers数量和突变的序列长度上一致。
其实这个亚基的保守性还是较高的,甚至可以把几个不一样的氨基酸手动突变一下工作量也不大。
这几种方法都可以将同源结构的氨基酸突变为自己的蛋白的氨基酸序列。
Refinement
可以对整条链进行一个refine。
Refine > Chain Refine。这个工具栏选项没有的话可以从Calculate - Module里打开。
接下来就可以等待它自动Refine直到显示Accet Refinement? Success出现,会给出评估结果,选择Accept或者Reject。绿色-黄色-橙色-红色代表由好到坏。
这个过程中是可以用鼠标挪动原子的,但是要注意温和一些,不然电脑可能会卡死。
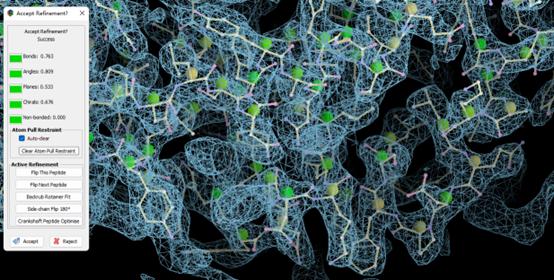
Refine这个过程有许多参数是可以调的。
比如:
-
点击右边工具栏的
R/RC可以调出Refine的参数,Refinement Weight可以调整Map权重。
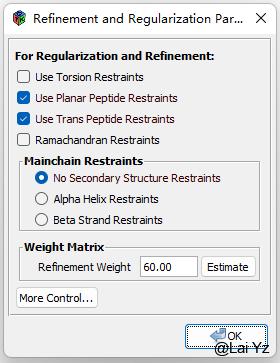
-
可以选择对原子添加约束。
Calculate - Module - Restraints - Generate All Molecule Self Restraints 5
这个一般错综复杂,可以将其隐藏。
Restraints - Undisplay Extra Distance Restraints
用完删除。
Restraints - Deletet All Extra Restraints
-
可以调整Geman-McClure约束的Alpha值(默认1)。
Refine - Set Geman-McClure alpha 0.1
-
对Map进行处理。
Calculate - Module - CryoEM
Cryo-EM - Sharpen/Blur - Resample - Make Map
尝试不同参数生成更加平滑的Map。
调节参数直到达到较好的效果。
不过一般情况下默认的Refine参数够用了。
工具栏的工具
在实际修正的过程中,右侧和上方工具栏的工具是经常使用到的。
Refine/Regularize/Control
点开 会弹出Refine参数,刚刚提到可以调节Refine中的Map weight。默认是带Planar Peptide和Trans Peptide约束的,比如在密度显示某处需要反转Trans-Cis Peptide的时候就可以把这个去掉,不然默认Refine是不太会自动帮你反转的。
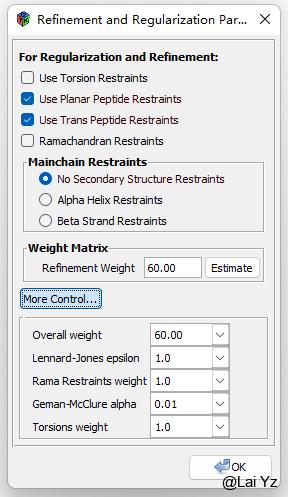
也可以按需要勾上Torsion Restraints和Ramachandran Restraints来添加侧链约束和主链约束,以免Refine的时候把已经修正好的地方给弄坏。这些的权重也是可以再More Control中调整的。
![]() 紫色的球是Real Space Refinement,
紫色的球是Real Space Refinement,![]() 白色的球是Regularize Zone。紫色的会根据一定的密度权重和一定的立体化学属性来进行Refine,白色的则是偏重蛋白质立体化学来把氨基酸骨架和侧链调整到一个科学的位置。
白色的球是Regularize Zone。紫色的会根据一定的密度权重和一定的立体化学属性来进行Refine,白色的则是偏重蛋白质立体化学来把氨基酸骨架和侧链调整到一个科学的位置。
它们都受到上面的参数影响。它们的使用方式都是点击球,然后点击两个原子,在两个原子所在残基之内的范围进行Refine。如果点击两次一个原子,就是只Refine这个残基。
Fix Atom
![]() 这是固定原子的工具,点击Fix Atom然后点击要固定的原子。在Refine的时候这些个原子就不会动了。
这是固定原子的工具,点击Fix Atom然后点击要固定的原子。在Refine的时候这些个原子就不会动了。
Rigid Movement
![]()
![]() 这两个都是将部分残基整体移动的,不会改变他们的几何构象,一个是平移,另一个还包括了旋转。
这两个都是将部分残基整体移动的,不会改变他们的几何构象,一个是平移,另一个还包括了旋转。
Rotamer
![]()
![]() 这两个是修正氨基酸侧链的立体几何的,会给出最合理的几种Rotamer并自己选择确认,
这两个是修正氨基酸侧链的立体几何的,会给出最合理的几种Rotamer并自己选择确认,![]() 绿色的会自动选择一种合适的Rotamer。这两个都不考虑密度而是考虑立体化学的合理性,因此密度好的地方还需要根据密度来酌情考量。
绿色的会自动选择一种合适的Rotamer。这两个都不考虑密度而是考虑立体化学的合理性,因此密度好的地方还需要根据密度来酌情考量。
Flip Peptide
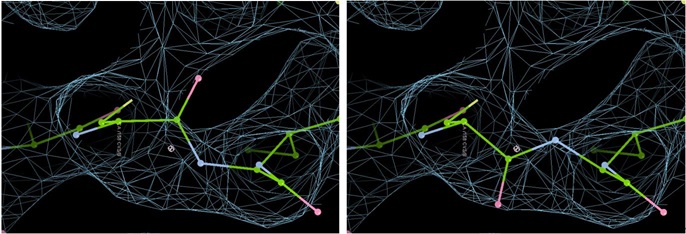
![]() 这个是翻转氨基酸主链构象的,有的地方主链搭错了,Ramachandron Plot显示异常,就有可能是主链反了180°,可以快速地通过这个功能翻转过来。
这个是翻转氨基酸主链构象的,有的地方主链搭错了,Ramachandron Plot显示异常,就有可能是主链反了180°,可以快速地通过这个功能翻转过来。
Flip Sidechain
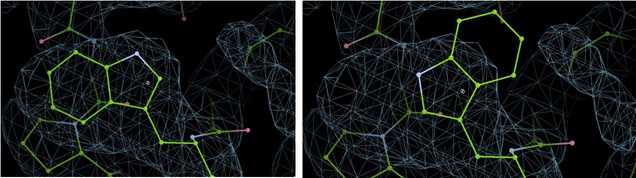
![]() 类似于Flip peptide,这个是翻转侧链构象180°。比如经常出现在Trp中,侧链较大且不对称,之前的文章提到过Phenix的Real Space Refinement对于这种大侧链的修正有时会有明显肉眼可见的错误。这种就可以通过Flip peptide快速翻转过来,不用自己手拖很费劲。
类似于Flip peptide,这个是翻转侧链构象180°。比如经常出现在Trp中,侧链较大且不对称,之前的文章提到过Phenix的Real Space Refinement对于这种大侧链的修正有时会有明显肉眼可见的错误。这种就可以通过Flip peptide快速翻转过来,不用自己手拖很费劲。
Change Residues phi and psi
![]() 这个功能可以纯手动调节氨基酸主链的phi和psi角,可操控性很高,所以需要丰富的经验,用的比较少。
这个功能可以纯手动调节氨基酸主链的phi和psi角,可操控性很高,所以需要丰富的经验,用的比较少。
Mutate
![]()
![]() 这两个是点突变氨基酸,可以突变单独一个氨基酸至20种常见氨基酸,区别在于一个只是突变,另一个在突变完后对氨基酸自动做一个Refine,实测效果常常一般,建议还是简单突变后手动修正。
这两个是点突变氨基酸,可以突变单独一个氨基酸至20种常见氨基酸,区别在于一个只是突变,另一个在突变完后对氨基酸自动做一个Refine,实测效果常常一般,建议还是简单突变后手动修正。
Add Residue
![]() 这个是在肽链的末端添加一个氨基酸,默认添加Alanine,如果点击中间的氨基酸是不会添加的,必需有一端是空的。
这个是在肽链的末端添加一个氨基酸,默认添加Alanine,如果点击中间的氨基酸是不会添加的,必需有一端是空的。
![]() 这个是给一个氨基酸添加两种不同的构象,用到的时候比较少。
这个是给一个氨基酸添加两种不同的构象,用到的时候比较少。
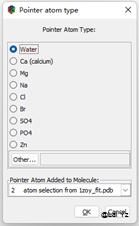
![]() 这个是在当前位置添加一个原子。包括常用的水分子和一些离子都可以用这个添加。
这个是在当前位置添加一个原子。包括常用的水分子和一些离子都可以用这个添加。
Delete Items
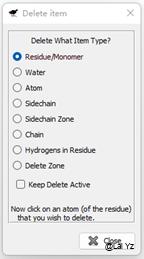
![]() 这个垃圾桶一样的工具是删除,可以选择删除残基、水分子、单个原子、侧链、整条链、氢原子以及某范围氨基酸肽链。默认是点击一下 再点一个对象就删除一个,如果勾上了Keep Delete Active,就可以鼠标左键一直点击删除,适合删除多个分子。
这个垃圾桶一样的工具是删除,可以选择删除残基、水分子、单个原子、侧链、整条链、氢原子以及某范围氨基酸肽链。默认是点击一下 再点一个对象就删除一个,如果勾上了Keep Delete Active,就可以鼠标左键一直点击删除,适合删除多个分子。
Undo & Redo
![]()
![]() 这个很好理解,撤回和重做。
这个很好理解,撤回和重做。
Manage Button
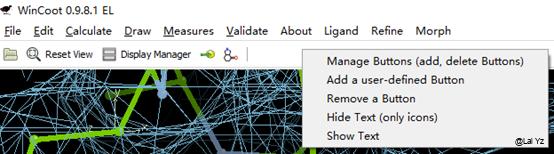
在上方工具栏中可以调出一些好用的工具,在工具栏点击鼠标右键 – Manage Buttons。
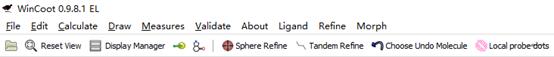
比如说Sphere Refine,可以对当前氨基酸为中心球形范围内的氨基酸进行Refine。
Tandem Refine则是对当前氨基酸前后几个氨基酸的一条链上进行Refine。
Choose Undo Molecule在打开了多了模型的时候会有用。
Local probe dots会显示附近的相互作用及Clash冲突。
还有许多其它的小工具可以自己添加。
评估
Coot有实时Validate的功能,可以一边修正一边看修正的效果。这些参数的具体意义可以参考之前关于氨基酸结构立体化学的基础文章。
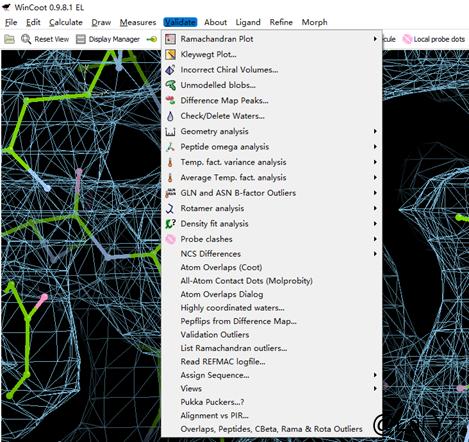
Ramachandron Plot
显示拉式图。Selection可以选择显示的范围,Outlier Only可以只显示Ramachandron outlier。点击小圆点就可以移动到对应的地方看看主链哪里不对劲。
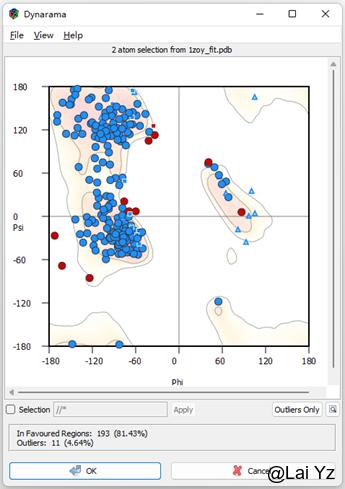
Geometry Analysis
Geometry会显示蛋白质一些Bond、Angle、Plane distortion等z score,可以参考看看。也是绿色较好,红色较差。
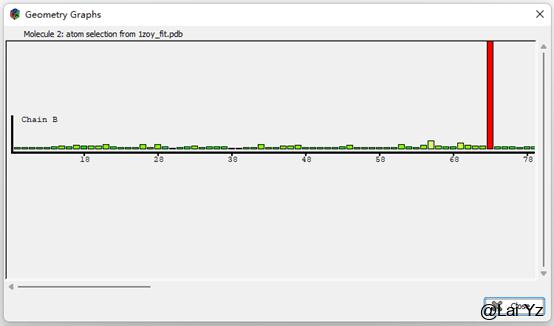
Peptide Omega Analysis
这是显示氨基酸Omega角度。
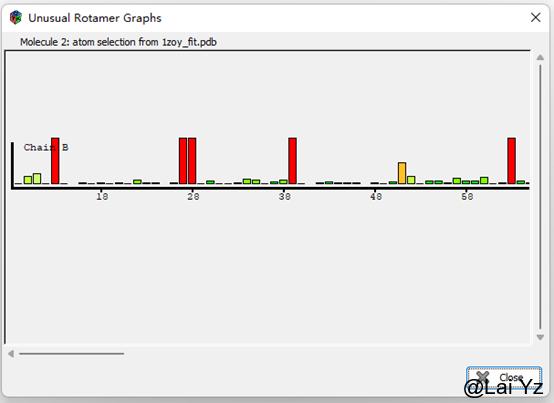
Rotamer Analysis
这是氨基酸侧链Rotamer outlier。
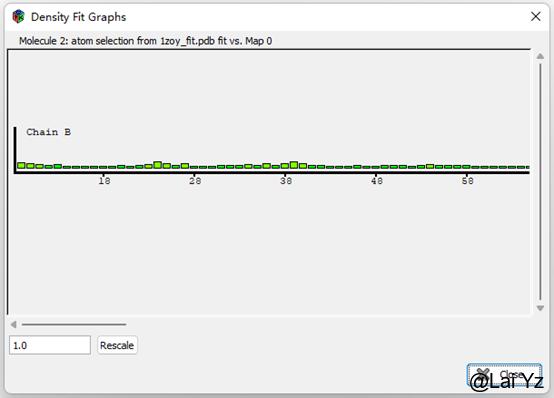
Density Fit Analysis
这是显示原子模型和密度匹配程度的。
Probe Clash
如果原子模型不带氢,会自动生成一个带氢的原子模型,然后计算Clash,并显示出来。红色的是Severe Clash。
Coot的Validate可以结合参考Phenix的Validation Report一起对原子模型进行修正。加上PDB数据库的Validate,这些都是对蛋白的立体化学性质等的评估,并不是绝对的,有的可能红色也能接受,需要综合考虑。
配体
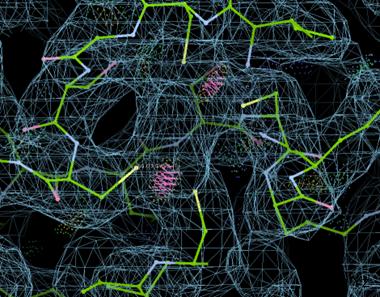
注意到chain B其实是有Ligands的,我们在Copy Fragment的时候特意没有添加进来。比如这个地方是有一个FES的密度的,与周围的四个Cys成键。
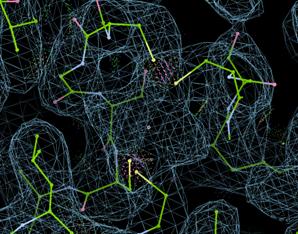
而有些Ligands在没有合适的限制文件的时候是无法Refine的,Coot不能识别。所以有时前期会先把Ligands去掉,后面在加上修正。
当然Coot本身也下载了一个基本化合物库,大部分情况下没有cif它也能识别大部分的配体并进行Refine,常见配体(包括FES)可以直接在Coot的File - Get Monomer直接导入,这里为了展示导入外源配体cif的方法。
比如先在Phenix中生成的二铁二硫铁硫簇FES的cif文件,点击File - Import CIF Dictionary,然后选择生成的cif限制文件,如果cif文件带原子坐标,可以勾上下面的Generate a Molecule,这样可以生成一个Ligand分子,否则需要再打开一个Ligand的原子坐标文件。
有了正确的cif,Coot才能识别并Refine分子。
感觉Rosseta的自动调整也还可以,还有个方便的gui RosEM